

Κρανιοσυνοστέωση: μια σπάνια νόσος που αντιμετωπίζεται
Το «κλειδί» για την αντιμετώπιση της κρανιοσυνοστέωσης, όπως και όλων των σπάνιων νόσων, είναι η ακριβής γενετική διάγνωση και η επιλογή του κατάλληλου σχεδίου θεραπείας από τους κατάλληλους ειδικούς.
«Τις εβδομάδες πριν από τη γέννηση του γιου μου Όλιβερ τον περασμένο Απρίλιο, το μόνο που μπορούσα να σκεφτώ ήταν η COVID-19. Η Ν. Υόρκη είχε εκατοντάδες θανάτους την ημέρα από τον ιό και έπρεπε να τηρηθούν τα πρωτόκολλα σχετικά με τον τοκετό. Όταν επιστρέψαμε στο σπίτι δυο μέρες μετά την καισαρική ένιωσα ανακούφιση.
Με
Αυτό που συνέβη στον Όλιβερ θα μπορούσε να συμβεί σε όλους μας. Ένα ατύχημα σαν αυτά που συμβαίνουν καθημερινά. Ένα ατύχημα στο γενετικό μας υλικό πριν από την γέννηση ή ένα αυτοκινητιστικό ατύχημα μετά την γέννηση. Ένα ατύχημα που προκαλεί αναπηρία ή δυσμορφία. Πώς θα μάς αντιμετώπιζαν τότε οι άλλοι, εμείς πώς θα αντιμετωπίζαμε την κατάστασή μας, αλλά και πώς αντιμετωπίζουμε γενικά αυτούς τους ανθρώπους;
Σήμερα υπολογίζεται ότι υπάρχει μια παγκόσμια κοινότητα περίπου 300 εκατομμυρίων ανθρώπων που ζουν με ένα σπάνιο νόσημα. Σε πανευρωπαϊκό επίπεδο, οι περίπου 6.500-8.000 καταγεγραμμένες ως σπάνιες νόσοι πλήττουν, με βάση εμπειρικές μελέτες, 37 εκατομμύρια άτομα του ευρωπαϊκού πληθυσμού. Στην Ελλάδα, με βάση και την ευρωπαϊκή εμπειρία, τα άτομα που πάσχουν από κάποια σπάνια νόσο υπολογίζονται περίπου σε 600.000.
Περίπου 5,2/10.000 γεννήσεις παιδιών-με μεγαλύτερη συχνότητα εμφάνισης στα αγόρια παρά στα κορίτσια- εμφανίζουν κρανιοσυνοστέωση, συχνά γενετικής αιτιολογίας. Μόνο το 2019, ο αριθμός των παιδιών στον κόσμο που γεννήθηκαν με κρανιοσυνοστέωση άγγιξε τις 84.665. Στην Ελλάδα τα άτομα με κρανιοσυνοστέωση διαφόρων τύπων υπολογίζονται σε περισσότερα από 4500. Σε ορισμένες περιπτώσεις, που ονομάζονται συνδρομικές, η νόσος σχετίζεται και με άλλες ανωμαλίες όπως τα σύνδρομα Apert ή Pfeiffer.
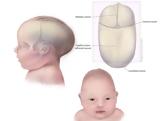
Η σπάνια κρανιοσυνοστέωσηΗ κρανιοσυνοστέωση συμπεριλαμβάνεται σε μια ομάδα γενετικών νοσημάτων, γνωστά ως σύνδρομα κεφαλής, που μελετώνται και στην Ελλάδα μαζί με τα κρανιοπροσωπικά και στοματοπροσωπικά σύνδρομα. Πρόκειται για μια γενετική ανωμαλία κατά την οποία μια ή περισσότερες ραφές στο κεφάλι ενός μωρού κλείνουν πρόωρα. Κανονικά, τα νεογέννητα έχουν «ανοικτές» ραφές που διαχωρίζουν τα οστά στο κρανίο τους δημιουργώντας τον απαραίτητο χώρο για να αναπτυχθεί φυσιολογικά ο εγκέφαλος. Πρόκειται για τα «μαλακά σημεία» που συνήθως αγχώνουν τους νέους γονείς αλλά που είναι ζωτικής σημασίας για την ταχεία ανάπτυξη του εγκεφάλου που συμβαίνει πριν από την ηλικία των 3 ετών.
Υπάρχουν διάφοροι τύποι κρανιοσυνοστέωσης ανάλογα με το ποιες εγκεφαλικές ραφές συγχωνεύονται, για τους περισσότερους από τους οποίους υπεύθυνες είναι μεταλλαγές στα γονίδια FGFR1, FGFR2, FGFR3, TWIST1 και MSX2, αλλά και σε αρκετά άλλα σπανιότερα γονίδια. Ο Όλιβερ που προαναφέρθηκε έπασχε από την πιο κοινή, την οβελιαία συνοστέωση (επίσης γνωστή ως σκαφοκεφαλία), που επηρεάζει την κύρια ραφή που εκτείνεται από το μπροστινό προς το πίσω μέρος του κρανίου και που αν δεν αντιμετωπιστεί, προκαλεί ένα ανώμαλο σχήμα κεφαλής που μπορεί να περιορίσει την ανάπτυξη του εγκεφάλου και να πιέζει το κρανίο. Μπορεί επίσης να προκαλέσει και ψυχοκοινωνικά προβλήματα καθώς το παιδί θα αλληλεπιδρά με τους συνομηλίκους του κατά την ανάπτυξη.
«Στις σπάνιες παθήσεις που μελετάμε περιλαμβάνονται οι κρανιοσυνοστεώσεις, οι στοματοπροσωπικές σχιστίες χείλους και υπερώας (περίπου 15000 άτομα στην Ελλάδα), οι διάφοροι τύποι εξωδερμικής δυσπλασίας (περίπου 1600 άτομα στην Ελλάδα), οι διάφοροι τύποι καρκίνου και κυρίως το ακανθοκυτταρικό καρκίνωμα στόματος (περίπου 550 άτομα στην Ελλάδα), αλλά και πολλά άλλα σπανιότερα γενετικά και πολυπαραγοντικά σύνδρομα (που υπολογίζονται συνολικά σε 3000 άτομα περίπου στην Ελλάδα), που συνήθως χρειάζονται ειδική διαγνωστική και θεραπευτική αντιμετώπιση», εξηγεί ο Αναπληρωτής Καθηγητής Γενετικής της Ιατρικής Σχολής του Εθνικού Καποδιστριακού Πανεπιστημίου Αθηνών Χρήστος Γιαπιτζάκης, ο οποίος μελετά τέτοια περιστατικά ως υπεύθυνος της ειδικής Μονάδας Στοματοπροσωπικής Γενετικής του Ερευνητικού Πανεπιστημιακού Ινστιτούτου Ιατρικής Ακριβείας και Υγείας Μητέρας και Παιδιού, με διευθυντή τον Ομότιμο Καθηγητή Γεώργιο Χρούσο.
O Αναπληρωτής Καθηγητής Γενετικής της Ιατρικής Σχολής του Εθνικού Καποδιστριακού Πανεπιστημίου Αθηνών Χρήστος Γιαπιτζάκης
Η Μονάδα Στοματοπροσωπικής Γενετικής της Ιατρικής Σχολής του ΕΚΠΑ που λειτουργεί τα τελευταία χρόνια στο Νοσοκομείο Παίδων «Αγία Σοφία», είναι η πρώτη εξειδικευμένη, και μέχρι τώρα παραμένει η μοναδική, που δημιουργήθηκε στον ελληνικό χώρο για την έρευνα, τη διαγνωστική μελέτη και τη διεπιστημονική αντιμετώπιση παρόμοιων σπάνιων περιστατικών με εκδηλώσεις από το στόμα, το πρόσωπο και την κεφαλή. Η νεοσύστατη αυτή μονάδα λειτουργεί με στόχο την κλινική και εργαστηριακή διάγνωση, τη γενετική συμβουλευτική σε οικογένειες και τον συντονισμό της θεραπευτικής αντιμετώπισης των καταστάσεων αυτών από μία ευρεία ομάδα ειδικών ιατρών και επιστημόνων.
Σύμφωνα με τον Καθηγητή Γιαπιτζάκη οι περισσότερες σπάνιες παθήσεις είναι γενετικής αιτιολογίας και στο 75% των περιπτώσεων εκδηλώνονται είτε κατά τη γέννηση, είτε κατά την παιδική ηλικία. Μόνο στο 25% των περιπτώσεων παρατηρείται έναρξη στην ενήλικη ζωή. «Δυστυχώς το 30% των πασχόντων πεθαίνει πριν να συμπληρώσει την ηλικία των πέντε ετών και πολλές φορές πριν καν γίνει ακριβής διάγνωση. Το ανησυχητικό είναι ότι σε αρκετές περιπτώσεις δε ζητείται η συμβολή ειδικών κλινικών γενετιστών και οι ασθενείς υποβάλλονται σε γενικές εξετάσεις, καθώς τα πρώτα συμπτώματα που είναι συνήθως κοινά με άλλες ασθένειες, οδηγούν σε γενικευμένες ή εσφαλμένες πρώτες διαγνώσεις. Έτσι, χωρίς την έγκαιρη ταυτοποίηση του ακριβούς σπάνιου νοσήματος, οι ασθενείς και οι οικογένειές τους ταλαιπωρούνται, ενώ καθυστερεί η κατάλληλη θεραπευτική αντιμετώπιση από τους κατάλληλους ανά περίπτωση ειδικούς. Η ακριβής γενετική διάγνωση ενός σπάνιου νοσήματος μπορεί να προσφέρει τη δυνατότητα της βέλτιστης θεραπευτικής αντιμετώπισης, αλλά και της στοχευμένης πρόληψης νέων παρόμοιων περιστατικών σε μια οικογένεια».
Αντιμετωπίζεται η κρανιοσυνοστέωση;Η διαχείριση και η θεραπεία των παιδιών με κρανιοσυνιοστέωση μπορεί να διαφέρουν ανάλογα με τον εντοπισμό και τη σοβαρότητα, την ηλικία και τις ανάγκες του παιδιού, αλλά και την παρουσία συνοδών συμπτωμάτων ή άλλων γενετικών ανωμαλιών. Ανάλογα με την περίπτωση και την ακριβή γενετική διάγνωση επιλέγεται το κατάλληλο σχέδιο θεραπείας από τους κατάλληλους ειδικούς.
Η τρέχουσα διαχείριση ασθενών με ενδογενή κρανιοσυνοστέωση είναι η χειρουργική διόρθωση. Η χειρουργική παρέμβαση για τη διαχείριση συγκεκριμένων εγκεφαλικών ραφών συνήθως επιδιώκεται πριν από την ηλικία του ενός έτους. Ενώ η συνδρομική κρανιοσυνοστέωση τυπικά απαιτεί πολύπλοκες σταδιακές κρανιακές και εξωκρανιακές επεμβάσεις, οι μη συνδρομικοί ασθενείς συνήθως χρειάζονται μόνο μία διαδικασία, χωρίς να αποκλείονται και περισσότερες. Αν και οι μη συνδρομικοί ασθενείς θα μπορούσαν πιθανώς να ζήσουν σχετικά φυσιολογική ζωή χωρίς χειρουργική επέμβαση, η χειρουργική διόρθωση εξακολουθεί να προτιμάται για αισθητικούς και αναπτυξιακούς λόγους.
Unsplash
Ωστόσο αυτό που πρέπει να τονιστεί είναι ότι η ορθή αντιμετώπιση της κρανιοσυνοστέωσης αποτελεί αποκλειστικά έργο μιας ομάδας ειδικών ιατρών και άλλων επιστημόνων και κυρίως γενετιστών και περιλαμβάνει κλινική και εργαστηριακή διάγνωση, γενετική συμβουλευτική, καθώς και ειδική θεραπευτική αντιμετώπιση ανά περίπτωση (που μπορεί να περιλαμβάνει νευροχειρουργική, γναθοχειρουργική, ορθοδοντική, παιδοδοντιατρική, παιδιατρική, παιδοχειρουργική, ορθοφωνητική και ψυχολογική θεραπεία).
«Η συμβολή του γενετιστή είναι καθοριστική τόσο στην κλινική διάγνωση, όσο και στην πρόληψη νέων αντίστοιχων βαρέων περιστατικών μέσα στην οικογένεια. Με τη σωστή διάγνωση και την κατάλληλη θεραπεία από την ομάδα ειδικών, τα περισσότερα παιδιά με σύνδρομα κεφαλής και στοματοπροσωπικά σύνδρομα, εφόσον δεν έχουν άλλα μείζονα προβλήματα υγείας, αντιμετωπίζονται καλά με υψηλά αποτελέσματα αποκατάστασης και έχουν μια φυσιολογική ζωή» καταλήγει ο Καθηγητής Γιαπιτζάκης, ο οποίος διαθέτει πολύχρονη εμπειρία στην έρευνα και στη γενετική διάγνωση σπανίων νοσημάτων που αντανακλάται σε πολλές πρωτοποριακές διεθνείς δημοσιεύσεις.
Ο ίδιος έχει πραγματοποιήσει προγεννητικούς ελέγχους αρκετών σπανίων στοματοπροσωπικών και νευρογενετικών νοσημάτων για πρώτη φορά στην Ελλάδα και ορισμένων νοσημάτων για πρώτη φορά παγκοσμίως, όπως για τη φυλοσύνδετη εξωδερμική δυσπλασία, το σύνδρομο Kennedy και τη σπαστική παραπληγία τύπου 3.
Η ερευνητική ομάδα του έχει τιμηθεί στο παρελθόν με το πρώτο βραβείο βασικής έρευνας για ένα πειραματικό μοντέλο καρκίνου του στόματος σε Παγκόσμιο Συνέδριο Ογκολογίας Στόματος στην Ολλανδία, ενώ το 2022 πήρε το πρώτο βραβείο βασικής έρευνας στο Πανελλήνιο Ιατρικό Συνέδριο για μία πρωτοποριακή μελέτη στο βασικοκυτταρικό καρκίνωμα.
#ΚΡΑΝΙΟΣΥΝΟΣΤΕΩΣΗ #ΝΟΣΟΣ #ΘΕΡΑΠΕΙΑ
- Δημοφιλέστερες Ειδήσεις Κατηγορίας Ειδήσεις
- Προειδοποίηση Παπαδόπουλου για μεγάλο σεισμό στην Ελλάδα: «Τραντάζει» τη χώρα μας το ρήγμα της Ανατολίας;
- Άνγκελα Μέρκελ: Με μαλλί που θυμίζει... Μπόρις Τζόνσον, ψωνίζει από τον φούρνο της γειτονιάς
- Οι επόμενες ημέρες θα είναι πολύ δύσκολες για αυτά τα ζώδια
- Σαρακοστή: Η ασθένεια που μας "απειλεί" την περίοδο της νηστείας
- Τραγωδία στα Ιωάννινα: 40χρονος βρέθηκε νεκρός στο σπίτι του
- Χειμερινές εκπτώσεις 2023: «Τόσα χρόνια στην αγορά, τέτοια κρίση δεν έχω ξαναζήσει»
- Μπουράκ Χακί για σεισμό στην Τουρκία: «Έχασα ανθρώπους, υπάρχει θυμός για τους υπευθύνους» (Video)
- Ο Μιλιβόγεβιτς σκέφτεται τον Ολυμπιακό
- Εμφάνιση αλά... Μπόρις Τζόνσον από την Άνγκελα Μέρκελ - Δείτε φωτογραφία
- Βορίδης για Πολάκη: «Το κόμμα του δεν έχει πάρει αποστάσεις, υπάρχει απλώς η παραπομπή του στο Πειθαρχικό»
- Δημοφιλέστερες Ειδήσεις Dikaiologitika
- Πυροβολισμοί με δύο τραυματισμούς κοντά σε δημοτικό σχολείο της Γερμανίας
- Το Market Pass άναψε «φωτιές» στα ΚΕΠ, «δεν γίνονται όλα με ένα κλικ» λένε οι εργαζόμενοι
- Μαρία Συρεγγέλα: Η Ελλάδα κάνει βήματα μπροστά και εφαρμόζει την ισότητα παντού
- Σακχαρώδης Διαβήτης: Αίτια και τα βασικά βήματα για τη ρύθμισή του
- Νέες εξελίξεις με την υγεία του Μικρούτσικου - Τι είπε η Κατερίνα Καινούργιου
- Καιρός: «Καμπανάκι» Αρναούτογλου για λασποβροχές τις επόμενες ώρες
- Ερευνητές συρρίκνωσαν το… μικροσκόπιο
- Διαζύγιο βόμβα στη showbiz: Χώρισε ο Γιώργος Μαυρίδης μετά από εννέα μήνες γάμου
- Κρανιοσυνοστέωση: μια σπάνια νόσος που αντιμετωπίζεται
- Μικρή παράταση στην υποβολή ΑΠΔ- Σήμερα η καταβολή ασφαλιστικών εισφορών
- Τελευταία Νέα Dikaiologitika
- Κρανιοσυνοστέωση: μια σπάνια νόσος που αντιμετωπίζεται
- Μαρία Συρεγγέλα: Η Ελλάδα κάνει βήματα μπροστά και εφαρμόζει την ισότητα παντού
- Το Market Pass άναψε «φωτιές» στα ΚΕΠ, «δεν γίνονται όλα με ένα κλικ» λένε οι εργαζόμενοι
- Πυροβολισμοί με δύο τραυματισμούς κοντά σε δημοτικό σχολείο της Γερμανίας
- Σακχαρώδης Διαβήτης: Αίτια και τα βασικά βήματα για τη ρύθμισή του
- Διαζύγιο βόμβα στη showbiz: Χώρισε ο Γιώργος Μαυρίδης μετά από εννέα μήνες γάμου
- Ερευνητές συρρίκνωσαν το… μικροσκόπιο
- Νέες εξελίξεις με την υγεία του Μικρούτσικου - Τι είπε η Κατερίνα Καινούργιου
- Μικρή παράταση στην υποβολή ΑΠΔ- Σήμερα η καταβολή ασφαλιστικών εισφορών
- Καιρός: «Καμπανάκι» Αρναούτογλου για λασποβροχές τις επόμενες ώρες
- Τελευταία Νέα Κατηγορίας Ειδήσεις
- Γεροντικό: Φραγμός στα πάθη
- Κατασχέθηκαν και καταστράφηκαν 47.124 τεμάχια απομιμητικών προϊόντων και επιβλήθηκαν πρόστιμα άνω των 200.000 ευρώ
- Μητσοτάκης για Πολάκη: «Οπως στρώνεις, κοιμάσαι»
- ΕΛΣΤΑΤ: Μείωση 1,2% του όγκου πωλήσεων στο λιανικό εμπόριο της χώρας τον Δεκέμβριο 2022
- Ποια είναι τα οφέλη του καφέ για την υγεία
- Απίστευτο: Ο Ερντογάν μοιράζει λεφτά σε παιδιά στο Αντιγιαμάν
- Σαρακοστή: Η ασθένεια που μας "απειλεί" την περίοδο της νηστείας
- Μεσολόγγι: Νεκρός άντρας σε τροχαίο – Εικόνες σοκ από το σημείο

